El pasado 16 de abril de 2014, se publicó el nuevo reglamento europeo sobre los ensayos clínicos de uso humano. Éste nace con el fin de dotar de más transparencia y accesibilidad a las investigaciones clínicas y ofrecer una mayor protección para los participantes, así como reducir los tiempos de evaluación de los ensayos clínicos. ¿Qué cambios lleva consigo este nuevo reglamento? ¿Cómo se van alcanzar dichos objetivos? ¿Estamos realmente preparados?
En el año 2001 se publicó la Directiva Europea 2001/20/CE, que establecía las bases para estandarizar las legislaciones de los Estados miembros de la Unión Europea (UE), en materia de ensayos clínicos.
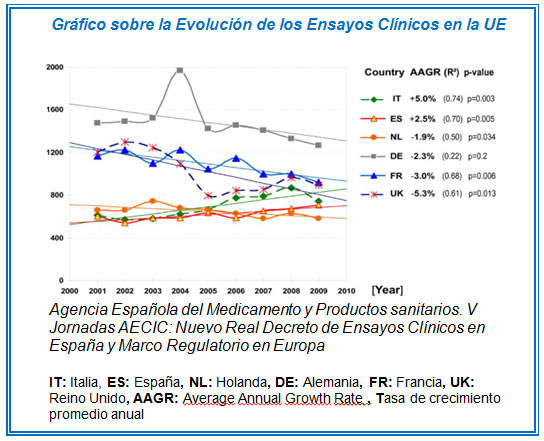
Cinco años después de su entrada en vigor, se observó que el número de ensayos clínicos realizados en la mayor parte de la UE disminuía (ver gráfico). Se despojaba así al “viejo continente” del papel preponderante que hasta entonces había ostentado.
Esta situación condujo a que, en el año 2009, la Agencia Europea de Medicamentos y la Federación Europea de Industrias Farmacéuticas iniciaran la redacción de un nuevo reglamento que marcara unas reglas de juego más claras: mayor transparencia y simplificación de los procedimientos, y mayor protección para los pacientes. El objetivo de estas mejoras era posicionar a la UE como el lugar idóneo para el desarrollo de ensayos clínicos.
El pasado mes de abril vio la luz el nuevo Reglamento Europeo, aunque no será hasta mayo del 2016 cuando entre en vigor.
Su publicación no ha dejado indiferente a nadie; alzando muchas voces críticas. Pero, ¿qué cambios llega consigo este nuevo reglamento?, ¿dotará de más transparencia y competitividad?
En este artículo se resumen los puntos clave sobre cómo esta legislación afectará en el día a día a la industria farmacéutica, Comités de ética, agencias europeas, pacientes, profesionales sanitarios, CROs y demás agentes implicados.
Búsqueda de una mayor transparencia en Investigación Clínica a través de:
* Bases de Datos
El nuevo reglamento establece la creación de una Base de Datos en la que se recoja toda la información relevante del ensayo clínico. Con esta Base de Datos, de acceso público, se pretende agilizar y facilitar el flujo de información entre promotores y Estados miembros.
La información recogida será, entre otra:
-
Resumen del protocolo, debe estar dirigido a personas legas en la materia,
-
Protocolo del ensayo,
-
Informe del estudio clínico,
-
Fechas de inicio y fin de la selección de los sujetos.
* Notificaciones
La nueva legislación amplia las notificaciones requeridas y las hace visibles para todos los Estados participantes en un mismo ensayo clínico. Los tiempos requeridos para realizarlas también varían:

*De manera general, las notificaciones descritas se realizarán en un plazo de 15 días a través del Portal Europeo, vía de comunicación entre las Agencias Europeas de Medicamentos (Estados miembros), los Comités de Ética y los promotores.
Alcanzar la simplificación de procedimientos y reducción de tiempos a través de:
* Evaluación del ensayo clínico
El nuevo reglamento trata de simplificar los procedimientos de aprobación de los ensayos clínicos, tanto para la autorización del ensayo como para la aprobación de las modificaciones, con el fin de ser más competitivos. A partir de mayo de 2016, toda solicitud será presentada a través de Portal único Europeo. Las evaluaciones se realizarán en dos partes:
-
Parte I: Evaluación conjunta realizada por los Estados miembros donde se vaya a llevar a cabo el ensayo clínico; actuando uno de ellos como Estado Miembro Notificante. Éste será el responsable de la preparación del informe preliminar y final.
-
Parte II: Evaluación independiente de cada Estado miembro, emitiendo cada uno un informe.
Los puntos a evaluar en la Parte I y Parte II se describen en la siguiente tabla:
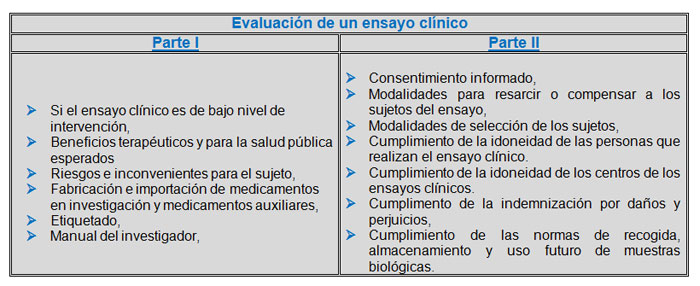
Para que el ensayo clínico sea autorizadolosinformes de ambas partes han de ser favorables.
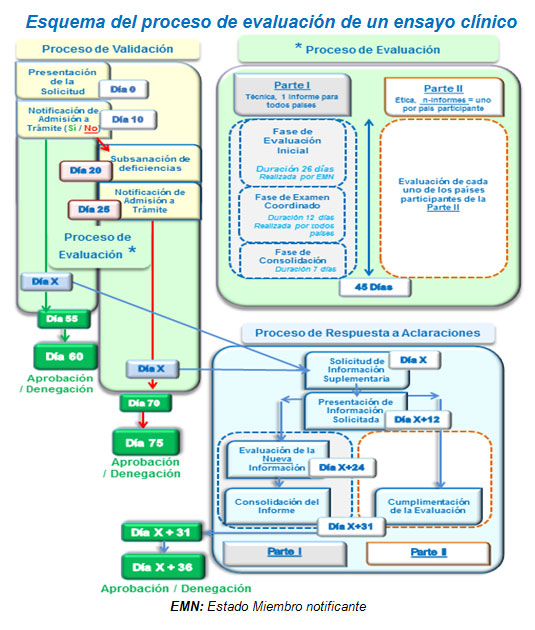
El proceso de evaluación de un ensayo clínico se llevará a cabo en un plazo mínimo de 60 días, siempre que la documentación presentada sea correcta y no se requieran aclaraciones. En el caso de requerirse aclaraciones, el plazo se verá ampliado añadiéndose 31 días desde la fecha en la que se notifiquen las mismas.
Actualmente los plazos vigentes de aprobación se sitúan entre 75 y 95 días, por lo que con la nueva legislación, en caso de requerirse aclaraciones, estos plazos no se verían reducidos.
* Modificaciones relevantes
Para la aprobación de las modificaciones relevantes (modificaciones sustanciales, según el nuevo Reglamento Europeo) que afecten a la Parte I, Parte II o ambas, el procedimiento es el mismo que el de la autorización de ensayo., El periodo de evaluación de estas modificaciones se reduce a 38 días, en comparación con los 35 días del actual Real Decreto. De este modo, la aprobación podría obtenerse en el plazo de 49 días, si la documentación presentada es conforme y no se requieren aclaraciones. Este plazo no resulta muy diferente del procedimiento actual (45 días).
* Validez de la autorización
Las autorizaciones de un ensayo clínico tendrán una validez de 2 años. Aquellos ensayos clínicos que tengan problemas de reclutamiento y prevean que, en este plazo, no se pueda incluir un paciente, han de solicitar una prórroga.
* Contratos con los centros
En este nuevo reglamento tampoco se establece cómo y cuándo se han de gestionar los contratos con los centros, punto crítico a la hora de iniciar los ensayos clínicos.
Aparición de nuevos escenarios y protección de los sujetos:
* Evaluadores del ensayo
Con el fin de garantizar la independencia, la transparencia, y una correcta evaluación del ensayo clínico, el reglamento establece en su artículo 9, que las evaluaciones se han de realizar por un número razonables de personas, las cuales:
-
Han de estar cualificadas mediante formación adicional y tener la experiencia necesaria,
-
No han de tener un conflicto de intereses, ni intereses financieros o personales que puedan afectar a su imparcialidad. Estas personas presentarán anualmente una declaración de intereses económicos,
-
Han de ser independientes al promotor y al centro de ensayo clínico, y de los investigadores implicados y de las personas que financien el ensayo clínico.
Al menos una de ellas ha de ser una persona lega en la materia y formará parte del Comité Ético.
El Comité Ético que evalúe el ensayo clínico no puede ser el del centro donde se vaya a desarrollar el ensayo.
* Consentimiento informado
Se incorpora un nuevo escenario, el Consentimiento Informado por grupos o simplificado, en el que únicamente se cumplimenta si el sujeto deniega su participación en el estudio.Para poder aplicar este tipo de consentimiento se ha de cumplir que:
-
no se contravenga la legislación del Estado miembro implicado.
-
el ensayo requiera establecergrupos de sujetos para administrar los diferentes medicamentos en investigación.
-
sea un ensayo clínico de bajo nivel de intervención y los medicamentos en investigación se usen bajo la indicación autorizada.
-
no se realicen intervenciones distintas a las del tratamiento estándar.
-
en el protocolo se justifique la obtención de este tipo del consentimiento informado y quede descrito el alcance de la información que se proporcionará así como los medios empleados para ello.
Como nota curiosa, el nuevo reglamento establece que en los casos en los que el paciente no sepa leer y escribir se permite el uso de medios alternativos (p.e., grabadoras de audio o vídeo)para la obtención del consentimiento.
* Nueva terminología
Se incorporan nuevos términos y conceptos:
* Ensayo clínico de bajo nivel de intervención: aquél que cumpla todas y cada una de las siguientes condiciones:
-
Los medicamentos en investigación estén autorizados
-
El protocolo establezca que:
-
los medicamentos investigados se utilicen según la indicación autorizada, o que
-
el uso de los medicamentos en investigación se base en pruebas y esté respaldado por datos científicos publicados sobre seguridad y eficacia en alguno de los Estados miembros implicados
-
Que los procedimientos complementarios de diagnóstico o seguimiento entrañen un riesgomínimo para los sujetos, comparado con el de la práctica clínica habitual en alguno de los Estados miembros implicados.
* Medicamento auxiliar: medicamento utilizado en un ensayo clínico según el protocolo, pero no como medicamento en investigación.
De forma adicional a los cambios presentados, el nuevo reglamento plantea también cambios en el etiquetado de los medicamentos en investigación, de los medicamentos auxiliares y de los placebos, cambios en el protocolo…
Con todo lo expuesto anteriormente surge la duda de si realmente este nuevo modelo, acorde con lo recogido en el Reglamento Europeo, simplifica los procedimientos de aprobación y acorta los plazos. Así mismo aparecen puntos controvertidos como la protección de los sujetos y la confidencialidad de los promotores.
Probablemente en el próximo mes de noviembre, cuando la AEMPS publique el borrador del nuevo Real Decreto de ensayos clínicos, quedarán resueltasmuchas de las dudas surgidas tras la lectura del nuevo Reglamento Europeo, aunque habrá que esperar al contenido publicado en el BOE.







